
Post Market Surveillance
Medical devices regulation
Post-Market Surveillance (PMS) is a requirement found in ISO 13485 and its chapter “measurements, analysis and improvement”, as well as in ISO 14971, and its chapter “post-production activities”. The subject is not new and has however been highlighted with MDR 2017/745 and its chapter VII.
Chapter VII of the Regulation lists three approaches:
- Post-market surveillance (PMS)
- Vigilance (materiovigilance for MDR and reactovigilance for IVDR)
- Market surveillance
Many of our customers have questions, lost in the twists and turns of acronyms: PMS, PMCF, PSUR, CEP, CER… This article will focus on PMS, but we will also briefly discuss clinical evaluation. Here is an overview to clarify these concepts, the differences, and the relationships between what is hidden behind these acronyms.
Post-Market Surveillance (PMS) of the MD includes three distinct but complementary components: post-marketing surveillance (PMS) of the MD itself, vigilance (materiovigilance), as well as market surveillance (by the competent authorities). The Regulation specifies the information that manufacturers and other economic operators must collect and report to the authorities, as well as the measures that the latter can adopt in the event of non-compliance.
Acronyms
- PMS: Post Market Surveillance
- PMSP: Post Market Surveillance Plan
- PMSR: Post Market Surveillance Report
- PSUR: Periodic Safety Update Report
- PMCF: Post Market Clinical Follow-up
- CEP: Clinical Evaluation Plan
- CER: Clinical Evaluation Report
References
- MDR 2017/745
- Chapter VII - Post-Market Surveillance, Vigilance And Market Surveillance
- Annex III - Technical Documentation On Post-Market Surveillance
- Annex XIV - Clinical Evaluation And Post-Market Clinical Follow-Up
- Guide MDCG 2020-7 Post-market clinical follow-up (PMCF) Plan Template - A guide for manufacturers and notified bodies
- Guide MDCG 2020-8 Post-market clinical follow-up (PMCF) Evaluation Report Template - A guide for manufacturers and notified bodies
- Guide MDCG 2020-10 Safety reporting in clinical investigations of medical devices under the Regulation (EU) 2017/745
- Guide MDCG 2022-21 Guidance On Periodic Safety Update Report (Psur) According To Regulation (Eu) 2017/745 (MDR)
- Guide MDCG 2023-3 Questions and Answers on vigilance terms and concepts as outlined in the Regulation (EU) 2017/745 on medical devices
- Guide MDCG 2024-1 Guidance on the vigilance system for CE-marked devices DSVG 00
- Guide MDCG 2024-1-1 DSVG 01 Devices for Cardiac Ablation
- Guide MDCG 2024-1-2 DSVG 02 Coronary Stents and associated delivery systems
- Guide MDCG 2024-1-3 DSVG 03 Cardiac Implantable Electronic Devices (CIEDs)
- Guide MDCG 2024-1-4 DSVG 04 Breast Implants
- MEDDEV 2.12-1 rev 8 Guidelines MD Vigilance system
- MEDDEV 2.12_2 Post Market Clinical Follow-up Studies
- ISO/TR 20416:2020 : Medical Device Post Market Surveillance for manufacturer
Post-Market Surveillance (PMS)
(MDR 2017/745 Chapter VII section 1, articles 83 to 86)
Manufacturers must design, establish, document, implement, maintain and update a post-market surveillance system whose requirements vary depending on the class and type of medical device (MD). The post-market surveillance system makes it possible to collect, record and analyze data on the quality, performance and safety of a MD throughout its life cycle. The PMS also makes it possible to draw conclusions from this data collection, and to apply preventive or corrective measures (CAPA) and monitor them.
Post-Market Surveillance Plan (PMSP)
The PMSP (article 84) is part of the technical documentation referred to in Annex II and required for all classes of devices. The requirements for the PMSP are given in Section 1 of Annex III.
Post-Market Surveillance Report (PMSR)
The PMSR (article 85) is required for Class I MDs and Class A and B MD-IVDs. This report summarizes the results and conclusions of the analysis of post-marketing surveillance data that were collected in within the framework of the post-market surveillance plan (PMSP). Update frequency: The PMSR is updated as necessary and made available to the competent authority.
Periodic Updated Safety Report (PSUR)
The PSUR (article 86) replaces the PMSR for class IIa, IIb and III MDs, and for class C and D MD-IVDs. Manufacturers establish a PSUR for each MD, and, where applicable, for each category or group of devices. This report summarizes the results and conclusions of the analysis of post-market surveillance data that were collected under the Post-Market Surveillance Plan (PMSP). The PSUR describes:
- the conclusions of the assessment of the benefit/risk ratio,
- the main findings of Post-Marketing Clinical Monitoring (SCAC) or Post-Marketing Performance Monitoring for DM-IVDs (SPAC),
- the sales volume of the DM,
- an estimate of the population
- and, if possible, the frequency of use of the DM.
Update frequency:
- As needed and at least every two years for class IIa MDs.
- and at least once a year for class IIb and III medical devices and class C and D IVD medical devices
The PSUR is part of the technical documentation referred to in Annexes II and III, and in Annex XIII section 2 for custom-made devices.
PMS reporting summary table
|
|
Class I | Class IIa non-implantable | Class IIb non-implantable | Class IIa implantable | Class III and IIb implantable | Custom made devices |
| Required documents |
PMSP PMSR |
PMSP PSUR |
PMSP PSUR |
PMSP PSUR |
PMSP PSUR |
PMSP PMSR or PSUR according to the classification |
| Frequency | When necessary | Every 2 years | Annualy | Every 2 years | Annualy | According to their MDR classification (Annex VIII) |
| EUDAMED | No | No | No | Yes | Yes | No |
| PMSR/PSUR first availability | 2 years from the device certification date. For newly certified devices, a PSUR is not required at the time of initial certification | 2 years from the device certification date. For newly certified devices, a PSUR is not required at the time of initial certification | One year from the device certification date. For newly certified devices, a PSUR is not required at the time of initial certification | 2 years after device certification date. For newly certified devices, a PSUR is not required at the time of initial certification | One year after device certification date. For newly certified devices, a PSUR is not required at the time of initial certification | One year (class lIb and IIl) or two years (class lla devices) when the first Statement of Conformity (DoC) for custom made devices is issued |
| NB / CA availability |
Made available |
Made available | Made available | Made available + EUDAMED | Made available + EUDAMED | N.A / Made available upon request |
| Data collection period | Aligned on the device certification date | Aligned on the device certification date or in agreed schedule with NB |
Aligned on the device certification date or in agreed schedule with NB
|
Aligned on the device certification date or in agreed schedule with NB | Aligned on the device certification date or in agreed schedule with NB | Aligned on the device certification date |
| End of PMSR/PSUR requirement | PMSR remains mandatory by the manufacturer until the end of the device lifetime defined in the technical documentation has been reached | PSUR remains mandatory by the manufacturer until the end of the device lifetime defined in the technical documentation has been reached | PSUR remains mandatory by the manufacturer until the end of the device lifetime defined in the technical documentation has been reached | PSUR remains mandatory by the manufacturer until the end of the device lifetime defined in the technical documentation has been reached | PSUR remains mandatory by the manufacturer until the end of the device lifetime defined in the technical documentation has been reached | PMSR/PSUR remains mandatory by the manufacturer until the end of the device lifetime defined in the technical documentation has been reached |
Vigilance (materiovigilance et reactovigilance)
Following notification of a serious incident, the manufacturer immediately carries out investigations relating to this incident and cooperates with the competent authorities and, where applicable, the ON (article 89 §1).
Member States take the necessary measures to ensure that any information concerning a serious incident or a security corrective measure is subject to a centralized evaluation at national level (article 89 §2). When several member states are concerned, the national authorities coordinate among themselves, under the direction of a national coordinating authority (article 89 §9 and 10). The competent authority monitors the investigation carried out by the manufacturer into a serious incident and may launch an independent investigation (article 89 §4). The manufacturer ensures that information relating to the corrective measures taken is brought without delay to the attention of users of the device in question by means of a safety notice previously submitted to the competent authority (article 89 §8) introduced in the electronic system relating to vigilance and post-marketing surveillance. The safety notice is drawn up in one or more official languages of the Union defined by the Member State in which the safety corrective measure is taken (article 89 §8).
The Commission and Member States monitor data from an electronic system to identify new security risks. If an unknown risk is discovered or the frequency of a known risk changes significantly, the competent authority informs the manufacturer, who must then take the necessary corrective measures. (section 90).
The Commission shall adopt implementing acts to define the modalities and procedural aspects for the application of the provisions relating to post-market surveillance (Article 91).
| Aspect | Details |
| Typology of incidents | Serious incidents and corrective measures for specific devices. |
| Notification and notices | Notification of serious incidents, safety notices, and periodic reports by manufacturers. |
| Notification forms | Standardized forms for electronic and non-electronic notification. |
| Notification Deadlines | Deadlines for notifying security corrective actions and for reporting, depending on the severity of the incident. |
| Information exchange forms | Harmonized forms for the exchange of information between competent authorities. |
| Coordination procedures | Designation of a coordinating competent authority and coordinated assessment procedures. |
These implementing acts are adopted according to the examination procedure set out in Article 114 §3.
The information collected in the electronic system for collecting and processing information relating to vigilance and post-marketing surveillance is made available to the ON, the competent authorities of the Member States and the Commission. The Commission ensures that healthcare professionals and the public have an appropriate level of access to this information. Are transmitted via this system:
- Manufacturer reports on serious incidents and safety corrective measures;
- Periodic summary reports established by manufacturers;
- Manufacturer trend reports;
- PSURs;
- Manufacturer safety notices;
- Information exchanged between the competent authorities of the Member States and between them and the Commission relating to corrective measures taken or envisaged by manufacturers and relating to the coordination of their assessments (article 92).
Market surveillance
(MDR 2017/745 Chapter VII section 3, articles 93 to 100).
National competent authorities shall appropriately monitor the compliance characteristics and performance of the devices. They shall draw up annual programs for surveillance activities and devote appropriate material and human resources to them, taking into account the corresponding European programme, provided for in Article 105. They shall draw up an annual summary of the results of their surveillance activities and update it. available to other competent authorities via the electronic system relating to market surveillance (Article 93). The competent authorities may confiscate, destroy or otherwise render unusable MDs that present an unacceptable risk or falsified MDs if they deem it necessary in the interests of protecting public health (Article 93 §5). After each of the inspections carried out as part of the conformity check, the competent authority draws up a report on the inspection findings regarding compliance with the applicable legal and technical requirements. It communicates the content of the report to the economic operator who was the subject of the inspection and gives him the opportunity to submit observations. The final inspection report is entered into the electronic market surveillance system. The competent authorities of the Member States shall coordinate their market surveillance activities, cooperate and share, also with the Commission, the related results, in order to ensure a high and harmonized level of market surveillance in all Member States (Article 93 §9).
Where the competent national authorities of a Member State have reason to believe that a device is likely to present an unacceptable risk to health or safety or does not comply with the requirements of the applicable Regulation, they carry out an assessment of the device concerned. Economic operators cooperate with the competent authorities (article 94).
If a medical device is deemed unsafe, authorities require the manufacturer and economic operators to take corrective action, and may limit or withdraw the product from the market. These actions must be proportional to the risk and promptly communicated to the European Commission and other Member States. If the measures are not implemented, the authorities may intervene further. In the event of disagreement between Member States or with the Commission on these measures, the Commission evaluates and decides on their justification (article 95).
Where a Member State or the Commission considers that the risk to health and safety presented by a device cannot be satisfactorily mitigated by the measures taken by the Member State(s) concerned, the Commission may take the necessary measures and duly justified to ensure the protection of health and safety (Article 96).
Where the competent authorities of a Member State conclude that a device does not comply with the requirements of the Regulation but does not present an unacceptable risk to health or safety, they require the economic operator concerned to implement a end the non-compliance in question within a reasonable period that they define. When the economic operator does not put an end to the non-compliance within the set deadline, the Member State concerned shall without delay take all appropriate measures to restrict or prohibit the making available of the product on the market, or so that it -it is recalled or withdrawn from the market. It shall notify these measures without delay to the Commission and the other Member States via the electronic system relating to market surveillance. The Commission may define appropriate measures to be taken by the competent authorities (Article 97).
Where a Member State considers that, to protect health and safety, the making available on the market or putting into service of a device or a specific category or group of devices should be prohibited, restricted or subject to special requirements, or that the device or category or group of devices in question should be withdrawn from the market or recalled, it may take any necessary and justified measures. The Member State notifies these measures to the Commission and to all other Member States via the electronic system relating to market surveillance. The Commission decides whether national measures are justified or not. When the assessment carried out by the Commission in consultation with the MDCG and, where applicable, the economic operators concerned, shows that the making available on the market or putting into service of a device or category or a specific group of devices should be prohibited, restricted or subject to special requirements, or that the device or category or group of devices in question should be withdrawn from the market or recalled in all Member States to protect health and safety patients the Commission may adopt implementing acts to take necessary and duly justified measures (Article 98).
The competent authorities of the Member States shall indicate the exact grounds on which the decisions they take in the context of market surveillance are based. When one of the above measures is addressed to a particular economic operator, he is informed of the decision and the remedies available to him. The economic operator must be able to present its observations to the competent authority within an appropriate time frame. The measures are immediately withdrawn or modified if the operator has taken effective corrective measures and the device now complies with the requirements of the Regulation (article 99).
Information relating to the surveillance activity and the MDs is transmitted via the electronic system relating to market surveillance to all relevant competent authorities and, where applicable, to the NB which issued the certificate. They are accessible to member states and the Commission. Information exchanged between Member States is not made public when market surveillance activities and cooperation between Member States could be affected (Article 100).
Post-market clinical Follow-up (PMCF)
(MDR 2017/745 Annex XIV Part B, article 61).
The PMCF is a clinical monitoring after CE marking consisting of collecting real-life clinical data, to confirm the performance and safety claims of a MD. The PMCF is part of the manufacturer's PMS. PMCF data is therefore used to feed the PMS. All devices associated with high risk to the patient must be subject to a PMCF. The establishment of a PMCF is imperative when:
- Available clinical data does not cover the lifespan of the DM.
- The clinical data at the time of CE marking cannot be extrapolated to the entire target population due in particular to more restrictive controlled clinical investigations.
- There are clinical risks not controlled in the risk analysis.
The PMCF is part of the technical documentation of the DM. Being a formal requirement of European regulations, the possible absence of a PMCF must be justified (MDR Annex II §6.1 d).
Clinical Evaluation
Article 61 §1 of MDR 271/745 indicates that manufacturers “must plan, carry out and document a clinical evaluation” to enable: Confirmation of compliance with relevant general safety and performance requirements... under normal conditions of intended use of the device, as well as assessment of undesirable side effects and acceptability of benefit-risk ratio. .. must be based on clinical data providing sufficient clinical evidence
Two documents will be generated during the clinical evaluation, the clinical evaluation plan (CEP) and the clinical evaluation report (CER) The clinical evaluation must be carried out as part of an application for the first CE marking, and is updated periodically (every 1 to 5 years depending on the risk class of the device and its history on the market). This makes it possible to verify its compliance with regulatory requirements throughout the life of the MD.
The Clinical Evaluation Plan (CEP)
Planning is essential to an effective clinical assessment process. To demonstrate that a device achieves the expected performance, clinical benefits and safety specifications, it is necessary to first identify what these parameters are. The manufacturer must then explain how these parameters can be measured or demonstrated (i.e. what are the associated parameters) and what an acceptable result looks like (i.e. benchmarks or performance measures) . To do this, it is necessary to start with a clear and sufficiently detailed description of the intended use of the device, as well as a specification of the standards of care for that intended use (otherwise known as “state of the art”).
The Clinical Evaluation Report (CER)
A clinical evaluation report (CER) documents the findings of a clinical evaluation and clinical evidence for a medical device. A CER may contain clinical data from:
- Clinical investigation of the device currently being evaluated.
- Clinical investigation or other studies reported in the scientific literature for an equivalent device.
- Peer-reviewed scientific literature reporting other clinical experiences with the device being evaluated or an equivalent device.
- Clinically relevant post-market surveillance (PMS) data with particular emphasis on post-market clinical monitoring (PMCF).
The CER aims to demonstrate that there is sufficient clinical evidence to confirm the safety and performance, including clinical benefits, of the device being evaluated when used as intended. CERs are required for all medical devices in the EU and are part of the technical documentation of the Medical Device Regulation MDR 2017/745.
Summary
A picture is worth a thousand words, so here is an illustration explaining the interrelationships between the different elements, from the Post-Marketing Clinical Follow-up Report (PMCF), to the Post-Market Surveillance Report (PMSR) or to the Periodic Safety Update Report (PSUR) , to the Clinical Evaluation Report (CER), and finally to the Technical File (TF). The elements fit together like a Russian doll.
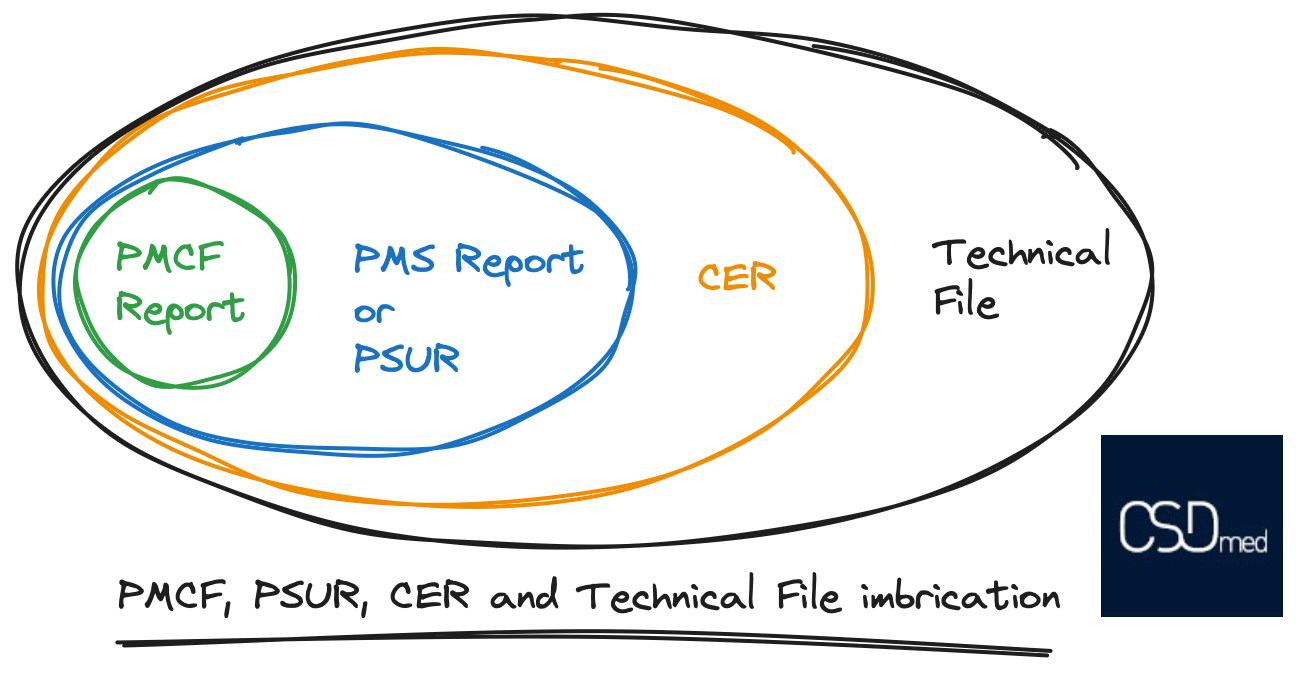
You can also refer to our article on the contents of the CE marking technical file.
We are at your service
European Regulation for Medical Devices MDR 2017/745 have added new requested requirements and these requirements form part of the documentation to be presented to the notified body (e.g. review of the in-depth clinical evaluation, PMS procedure, QC results from validation products, proof of staff skills, etc.).
CSDmed brings its expertise and a methodical approach to its clients, start-ups, manufacturers, importers and distributors of medical devices, thanks to a team of specialized experts and consultants, who will be able to address the MDR dossiers in their entirety
🔗 Contact us and find out how we can help you.