
MDCG 2023-7 Orientations sur les exemptions à l'obligation de réaliser des investigations cliniques conformément à l'article 61, paragraphes 4 à 6 du MDR et sur les niveaux d'accès suffisants aux données nécessaires pour justifier les allégations d'équiv
Reglementation des dispositifs medicaux
MDCG 2023-7 Orientations sur les exemptions à l'obligation de réaliser des investigations cliniques conformément à l'article 61, paragraphes 4 à 6 du MDR et sur les niveaux d'accès suffisants aux données nécessaires pour justifier les allégations d'équivalence.
Ces lignes directrices visent à clarifier les exemptions à l'obligation de réaliser des investigations cliniques, et les conditions associées liées à la démonstration de l'équivalence, pour les dispositifs médicaux implantables et de classe III destinés à être mis sur le marché européen. Il fournit également des exemples et des considérations pertinentes pour la démonstration de « niveaux suffisants d'accès aux données » conformément à la section 3 de l'annexe XIV.
Que peut-on trouver dans ce guide ?
Le document précise les conditions spécifiques dans lesquelles les investigations cliniques sur les dispositifs médicaux pourraient être exemptées. Par exemple, si un nouveau dispositif médical est similaire à un dispositif existant et approuvé, le fabricant peut revendiquer l’équivalence et potentiellement contourner des essais cliniques supplémentaires. Cette équivalence doit être justifiée par des données démontrant une similitude sur les plans technique, biologique et clinique. Il comprend des scénarios dans lesquels un fabricant compare son nouvel appareil à un appareil existant qu'il produit ou à l'appareil d'un concurrent, à condition qu'il dispose d'un accès adéquat aux données nécessaires. Le document souligne l'importance de justifier minutieusement toute allégation d'équivalence, en garantissant que la sécurité des patients et l'efficacité du dispositif ne soient pas compromises par l'exemption des essais cliniques.
4 scénarios
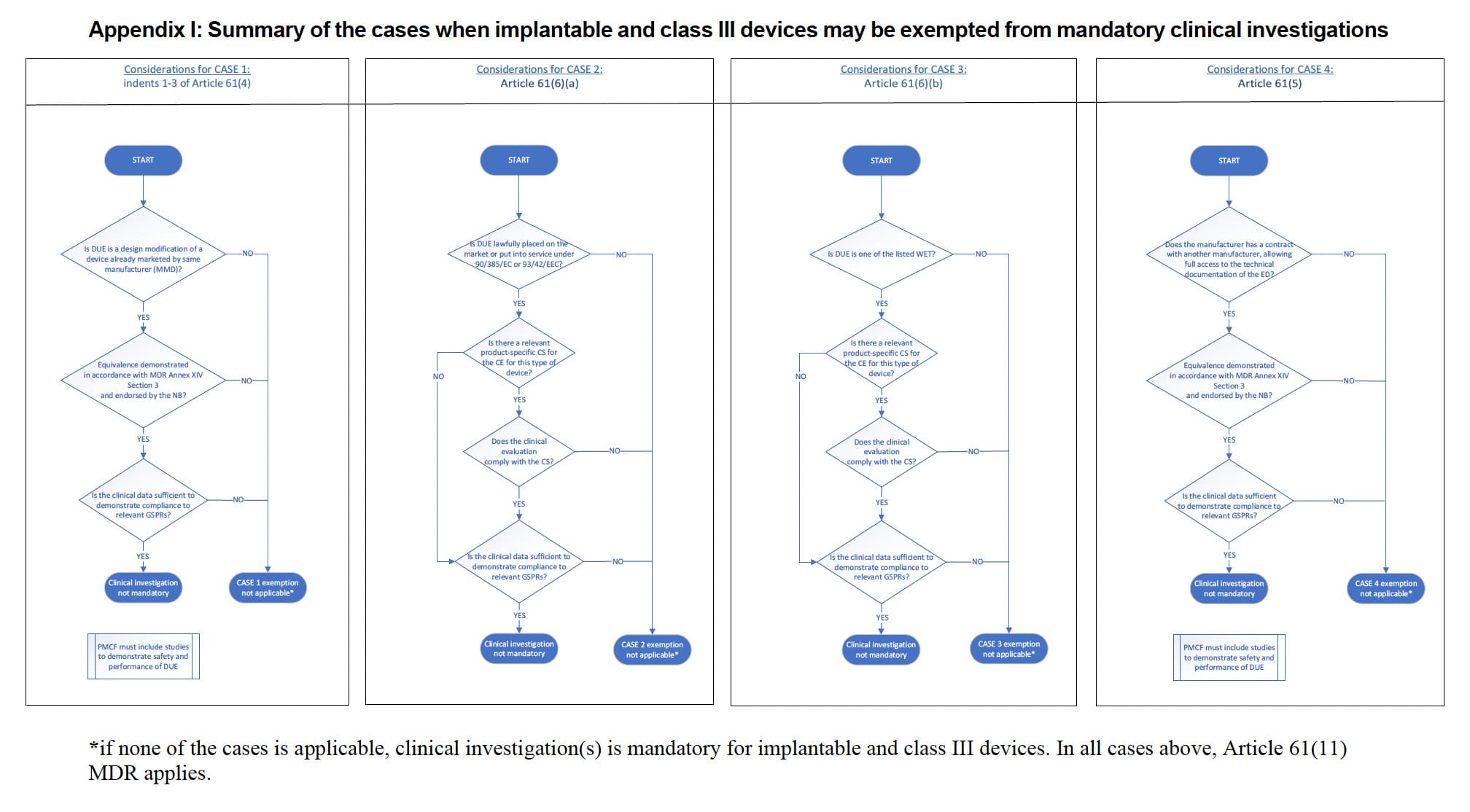
La ligne directrice MDCG 2023-7 décrit dans le tableau 1 quatre scénarios dans lesquels des exemptions aux investigations cliniques sont possibles :
Cas 1 : alinéas 1 à 3 de l’article 61, paragraphe 4 :
- Le dispositif en cours d'évaluation (DUE - Device under evaluation) a été conçu en modifiant un dispositif déjà commercialisé par le même fabricant.
- L’équivalence est démontrée entre le DUE et l’ED (Equivalent Device) du fabricant conformément au point 3 de l’annexe XIV ; la démonstration de l'équivalence a été approuvée par l'organisme notifié. Pour plus d’informations sur la démonstration de l’équivalence, veuillez vous référer au MDCG 2020-5.
- L'évaluation clinique du dispositif commercialisé est suffisante pour démontrer la conformité du dispositif modifié aux exigences de sécurité et de performance applicables.
- Le plan PMCF est approprié et comprend des études post-commercialisation pour démontrer la sécurité et les performances du DUE.
Cas 2 : article 61, paragraphe 6, point a :
- Le DUE a été légalement mis sur le marché ou mis en service conformément à la directive 90/385/CEE ou à la directive 93/42/CEE.
- L'évaluation clinique est basée sur des données cliniques suffisantes.
- L'évaluation clinique est conforme au CS spécifique au produit pertinent pour l'évaluation clinique de ce type de dispositif, lorsqu'un tel CS est disponible.
Cas 3 : Article 61(6)(b) :
- DUE fait partie des types de dispositifs répertoriés : « sutures, agrafes, obturations dentaires, appareils dentaires, couronnes dentaires, vis, cales, plaques, fils, broches, clips ou connecteurs ».
- L'évaluation clinique est basée sur des données cliniques suffisantes.
- L'évaluation clinique est conforme au CS spécifique au produit pertinent pour l'évaluation clinique de ce type de dispositif, lorsqu'un tel CS est disponible.
Cas 4 : article 61, paragraphe 5 :
- L’équivalence est démontrée entre le DUE et l’ED de l’autre fabricant conformément au point 3 de l’annexe XIV.
Le document comprend deux annexes :
1️⃣ L'Annexe I récapitule les cas où les dispositifs implantables et de classe III peuvent être exemptés des investigations cliniques obligatoires. Il détaille les scénarios spécifiques dans lesquels ces exemptions s'appliquent, aidant ainsi les fabricants à déterminer si leur appareil est admissible à une exemption.
2️⃣ L'Annexe II présente une hiérarchie des niveaux d'accès aux données concernant les caractéristiques cliniques, techniques et biologiques pour démontrer l'équivalence. Cette annexe fournit des exemples et suggère une hiérarchie liée au niveau d'accès aux données, indiquant les limitations potentielles et la manière dont elles pourraient être résolues.
Ces annexes sont cruciales pour comprendre les conditions dans lesquelles les investigations cliniques pour certains dispositifs médicaux peuvent être exemptées et comment justifier les demandes d'équivalence basées sur le niveau d'accès aux données.
Liens vers le document ci-dessous. Profitez-en, enjoy 💕
Nous sommes à votre service
Depuis l’arrivée du Règlement 2017/745, de nouvelles exigences sont demandées et font partie de la documentation à présenter à l’organisme notifié (par exemple revue de l’évaluation clinique poussée, procédure de PMS, résultats de CQ issus de la validation des produits, preuves de compétences du personnel, etc.).
CSDmed apporte son expertise et une approche méthodique à ses clients, start-ups, fabricants, importateurs et distributeurs de dispositifs médicaux, grâce à une équipe d’experts et de consultants spécialisés, qui pourront traiter la transition MDR dans son entièreté.
🔗 Contactez-nous et découvrez comment nous pouvons vous aider.