
La Surveillance Après Commercialisation
Reglementation des dispositifs medicaux
La surveillance après commercialisation (PMS) est une exigence que l’on retrouve dans l’ISO 13485 et son chapitre “mesures, analyse et amélioration”, ainsi que dans l’ISO 14971, et son chapitre “activités de post production”. Le sujet n’est pas nouveau et a cependant été mis en avant avec le MDR 2017/745 et son chapitre VII.
Le chapitre VII des Règlements liste trois approches :
- la surveillance après commercialisation (Post-market surveillance - PMS)
- la vigilance (matériovigilance pour le MDR (et réactovigilance pour l’IVDR) (Vigilance)
- la surveillance du marché (Market surveillance)
Beaucoup de nos clients se posent des questions, perdus dans les méandres des acronymes : PMS, PMCF, PSUR, CEP, CER… Cet article se concentrera sur la PMS, mais nous discuterons également brièvement de l'évaluation clinique. Voici donc un tour d’horizon pour clarifier ces concepts, les différences, et les relations entre ce qui se cahce derrière ces acronymes.
La surveillance post-commercialisation (PMS) du DM comprend trois volets distincts mais complémentaires : la surveillance après commercialisation (PMS) du DM en elle-même, la vigilance (matériovigilance), ainsi que la surveillance du marché (par les autorités compétentes). Le Règlement précise les informations que les fabricants et les autres opérateurs économiques doivent collecter et rapporter aux autorités, ainsi que les mesures que celles-ci peuvent adopter en cas de non-conformité.
Les acronymes
- PMS: Post Market Surveillance
- PMSP: Post Market Surveillance Plan ou Plan de Surveillance Après Commercialisation
- PMSR: Post Market Surveillance Report ou Rapport de Surveillance Après Commercialisation
- PSUR: Periodic Safety Update Report
- PMCF: Post Market Clinical Follow-up ou Suivi Clinique Après Commercialisation (SCAC)
- CEP: Clinical Evaluation Plan
- CER: Clinical Evaluation Report
Les références
- MDR 2017/745
- Chapter VII - Post-Market Surveillance, Vigilance And Market Surveillance
- Annex III - Technical Documentation On Post-Market Surveillance
- Annex XIV - Clinical Evaluation And Post-Market Clinical Follow-Up
- Guide MDCG 2020-7 Post-market clinical follow-up (PMCF) Plan Template - A guide for manufacturers and notified bodies
- Guide MDCG2020-8 Post-market clinical follow-up (PMCF) Evaluation Report Template - A guide for manufacturers and notified bodies
- Guide MDCG2020-10 Safety reporting in clinical investigations of medical devices under the Regulation (EU) 2017/745
- Guide MDCG 2022-21 Guidance On Periodic Safety Update Report (Psur) According To Regulation (Eu) 2017/745 (Mdr)
- Guide MDCG2023-3 Questions and Answers on vigilance terms and concepts as outlined in the Regulation (EU) 2017/745 on medical devices
- Guide MDCG2024-1 Guidance on the vigilance system for CE-marked devices DSVG 00
- Guide MDCG2024-1-1 DSVG 01 Devices for Cardiac Ablation
- Guide MDCG2024-1-2 DSVG 02 Coronary Stents and associated delivery systems
- Guide MDCG2024-1-3 DSVG 03 Cardiac Implantable Electronic Devices (CIEDs)
- Guide MDCG2024-1-4 DSVG 04 Breast Implants
- MEDDEV 2.12-1 rev 8 Guidelines MD Vigilance system
- MEDDEV 2.12_2 Post Market Clinical Follow-up Studies
- ISO/TR 20416:2020 : Medical Device Post Market Surveillance for manufacturer
La surveillance après commercialisation (PMS)
(MDR 2017/745 Chapitre VII section 1, articles 83 à 86)
Les fabricants doivent concevoir, établir, documenter, appliquer, maintenir et mettre à jour un système de surveillance après commercialisation dont les exigences varient selon la classe et le type de dispositif médical (DM). Le système de surveillance après commercialisation permet de collecter, enregistrer et analyser les données sur la qualité, les performances et la sécurité d’un DM pendant toute sa durée de vie. La PMS permet également de tirer les conclusions de cette collecte de données, et d’appliquer les mesures préventives ou correctives (CAPA) et d’en assurer le suivi.
Le Plan de Surveillance Après Commercialisation (PMSP)
Le PMSP (article 84) fait partie de la documentation technique visée à l’Annexe II et exigé pour toutes les classes de dispositifs. Les exigences pour le PMSP sont données en Section 1 de l’Annexe III.
Le Rapport de Surveillance Après Commercialisation (PMSR)
Le PMSR (article 85) est exigé pour les DM de classe I et les DM-DIV de classe A et B. Ce rapport établit la synthèse des résultats et des conclusions de l'analyse des données de surveillance après commercialisation qui ont été collectées dans le cadre du plan de surveillance après commercialisation (PMSP). Fréquence de mise à jour : Le PMSR est mis à jour selon les besoins et mis à disposition de l’autorité compétente.
Le Rapport Périodique Actualisé de Sécurité (PSUR)
Le PSUR (article 86) remplace le PMSR pour les DM de classe IIa, IIb et III, et pour les DM-DIV de classe C et D. Les fabricants établissent un PSUR pour chaque DM, et, le cas échéant, pour chaque catégorie ou groupe de dispositifs. Ce rapport fait la synthèse des résultats et des conclusions de l'analyse des données de surveillance après commercialisation qui ont été collectées dans le cadre du plan de surveillance après commercialisation (PMSP). Le PSUR décrit :
- les conclusions de l’évaluation du rapport bénéfice/risque,
- les principales constatations du Suivi Clinique Après Commercialisation (SCAC) ou du Suivi Des Performances Après Commercialisation pour les DM-DIV (SPAC),
- le volume des ventes du DM,
- une estimation de la population
- et, si possible, la fréquence d’utilisation du DM.
Fréquence de mise à jour :
- Selon les besoins et au moins tous les deux ans pour les DM de classe IIa.
- et au moins une fois par an pour les DM des classes IIb et III et les DM DIV de classe C et D
Le PSUR fait partie de la documentation technique visée aux annexes II et III, et à l’annexe XIII section 2 pour les dispositifs sur-mesure.
Table de synthèse des rapports PMS
|
|
Classe I | Classe lla non-implantable | Classe Ilb non-implantable | Classe lla implantable | Classe Ill and Ilb implantable | Dispositifs sur mesure |
| Documents exigés |
PMSP PMSR |
PMSP PSUR |
PMSP PSUR |
PMSP PSUR |
PMSP PSUR |
PMSP PMSR ou PSUR selon la classification MDR (Annexe VIII) |
| Fréquence de mise à jour | Lorsque nécessaire | Tous les 2 ans | Tous les ans | Tous les 2 ans | Tous les ans | Selon la classification MDR (Annexe VIII) |
| EUDAMED | Non | Non | Non | Oui | Oui | Non |
| Première version du PMSR/PSUR | 2 ans à partir de la date de certification du DM. Pour les DM nouvellement certifiés, le PSUR n’est pas exigé lors de la demande de certification initiale. | 2 ans à partir de la date de certification du DM. Pour les DM nouvellement certifiés, le PSUR n’est pas exigé lors de la demande de certification initiale. | 1 an à partir de la date de certification du DM. Pour les DM nouvellement certifiés, le PSUR n’est pas exigé lors de la demande de certification initiale. | 1 an à partir de la date de certification du DM. Pour les DM nouvellement certifiés, le PSUR n’est pas exigé lors de la demande de certification initiale. | 1 an à partir de la date de certification du DM. Pour les DM nouvellement certifiés, le PSUR n’est pas exigé lors de la demande de certification initiale. | 1 an (classe lIb et IIl) ou 2 ans (classe lla) à partir de la première Déclaration Européenne de Conformité (DoC) du DM sur mesure. |
| Mise à disposition pour les NB / CA | Mettre à disposition sur demande | Mis à disposition | Mis à disposition | Mis à disposition + EUDAMED | Mis à disposition + EUDAMED | N.A / Mettre à disposition sur demande |
| Période de collecte des données | Aligné avec la date de marquage CE | Aligné avec la date de certification du DM ou calendrier en accord avec l’ON. | Aligné avec la date de certification du DM ou calendrier en accord avec l’ON. | Aligné avec la date de certification du DM ou calendrier en accord avec l’ON. | Aligné avec la date de certification du DM ou calendrier en accord avec l’ON. | Aligné avec la date de certification du DM |
| Fin des exigences en PMSR/PSUR | Le PMSR reste obligatoire pour le fabricant jusqu’à ce que la fin de vie du DM, telle que définie dans le dossier technique, ait été atteinte. | Le PSUR reste obligatoire pour le fabricant jusqu’à ce que la fin de vie du DM, telle que définie dans le dossier technique, ait été atteinte. | Le PSUR reste obligatoire pour le fabricant jusqu’à ce que la fin de vie du DM, telle que définie dans le dossier technique, ait été atteinte. | Le PSUR reste obligatoire pour le fabricant jusqu’à ce que la fin de vie du DM, telle que définie dans le dossier technique, ait été atteinte. | Le PSUR reste obligatoire pour le fabricant jusqu’à ce que la fin de vie du DM, telle que définie dans le dossier technique, ait été atteinte. | Le PMSR ou PSUR reste obligatoire pour le fabricant jusqu’à ce que la fin de vie du DM, telle que définie dans le dossier technique, ait été atteinte. |
La vigilance (matériovigilance et réactovigilance)
(MDR 2017/745 Chapitre VII section 2, articles 87 à 92)
A la suite de la notification d’un incident grave, le fabricant mène sans tarder les investigations liées а cet incident et coopère avec les autorités compétentes et, le cas échéant, l’ON (article 89 §1).
Les Etats membres prennent les mesures nécessaires pour que toute information concernant un incident grave ou une mesure corrective de sécurité fasse l’objet d’une évaluation centralisée au niveau national (article 89 §2). Lorsque plusieurs Etats membres sont concernés, les autorités nationales se coordonnent entre elles, sous la direction d’une autorité nationale coordinatrice (article 89 §9 et 10). L’autorité compétente assure le suivi de l’investigation menée par le fabricant sur un incident grave et peut lancer une enquête indépendante (article 89 §4). Le fabricant veille à ce que les informations relatives aux mesures correctives prises soient portées sans tarder а l'attention des utilisateurs du dispositif en question au moyen d'un avis de sécurité préalablement soumis à l’autorité compétente (article 89 §8) introduit dans le système électronique relatif à la vigilance et à la surveillance après commercialisation. L'avis de sécurité est rédigé dans une ou des langues officielles de l'Union définies par l'état membre dans lequel la mesure corrective de sécurité est prise (article 89 §8).
La Commission et les États membres surveillent les données d'un système électronique pour identifier de nouveaux risques de sécurité. Si un risque inconnu est découvert ou la fréquence d'un risque connu change significativement, l'autorité compétente informe le fabricant, qui doit alors prendre les mesures correctives nécessaires. (article 90).
La Commission prend des actes d’exécution afin de définir les modalités et les aspects procéduraux pour l’application des dispositions relatives à la surveillance après la commercialisation (article 91).
| Aspect | Détails |
| Typologie des incidents | Incidents graves et mesures correctives pour des dispositifs spécifiques. |
| Notification et avis | Notification des incidents graves, avis de sécurité, et rapports périodiques par les fabricants. |
| Formulaires de notification | Formulaires standardisés pour la notification électronique et non électronique. |
| Délais de notification | Délais pour notifier les mesures correctives de sécurité et pour les rapports, en fonction de la sévérité de l'incident. |
| Formulaires d'échange d'informations | Formulaires harmonisés pour l'échange d'informations entre autorités compétentes. |
| Procédures de coordination | Désignation d'une autorité compétente coordonnatrice et procédures d'évaluation coordonnée. |
Ces actes d'exécution sont adoptés selon la procédure d'examen énoncée à l'article 114 §3.
Les informations recueillies dans le système électronique de collecte et traitement des informations relatives à la vigilance et à la surveillance après commercialisation sont mises à la disposition de l’ON, des autorités compétentes des états membres et de la Commission. La Commission veille à ce que les professionnels de santé et le public aient un niveau d’accès approprié à ces informations. Sont transmis via ce système :
- Les rapports des fabricants sur les incidents graves et les mesures correctives de sécurité ;
- Les rapports de synthèse périodiques établis par les fabricants ;
- Les rapports de tendances des fabricants ;
- Les PSUR ;
- Les avis de sécurité des fabricants ;
- Les informations échangées entre les autorités compétentes des Etats membres et entre celles-ci et la Commission relatives aux mesures correctives prises ou envisagées par les fabricants et relatives à la coordination de leurs évaluations (article 92).
La surveillance du marché
(MDR 2017/745 Chapitre VII section 3, articles 93 à 100).
Les autorités nationales compétentes contrôlent de manière appropriée les caractéristiques et les performances des dispositifs en matière de conformité. Elles élaborent des programmes annuels pour les activités de surveillance et y consacrent les ressources matérielles et humaines appropriées en tenant compte du programme européen correspondant, prévu à l’article 105. Elles établissent un résumé annuel des résultats de leurs activités de surveillance et le mettent à la disposition des autres autorités compétentes via le système électronique relatif à la surveillance du marché (article 93). Les autorités compétentes peuvent confisquer, détruire ou rendre inutilisables par d'autres moyens les DM qui présentent un risque inacceptable ou les DM falsifiés si elles le jugent nécessaire dans l'intérêt de la protection de la santé publique (article 93 §5). Après chacune des inspections effectuées dans le cadre du contrôle de la conformité, l'autorité compétente établit un rapport sur les conclusions de l'inspection concernant le respect des exigences légales et techniques applicables. Elle communique la teneur du rapport à l'opérateur économique qui a fait l'objet de l'inspection et lui donne la possibilité de présenter des observations. Le rapport d'inspection définitif est introduit dans le système électronique relatif à la surveillance du marché. Les autorités compétentes des Etats membres coordonnent leurs activités de surveillance du marché, coopèrent et partagent, également avec la Commission, les résultats y afférents, afin d'assurer un niveau élevé et harmonisé de surveillance du marché dans l'ensemble des états membres (article 93 §9).
Lorsque les autorités nationales compétentes d'un Etat membre ont des raisons de croire qu'un dispositif est susceptible de présenter un risque inacceptable pour la santé ou la sécurité ou n’est pas conforme aux exigences du Règlement applicable, elles réalisent une évaluation du dispositif concerné. Les opérateurs économiques coopèrent avec les autorités compétentes (article 94).
Si un dispositif médical est jugé dangereux, les autorités exigent du fabricant et des opérateurs économiques qu'ils prennent des mesures correctives, et peuvent limiter ou retirer le produit du marché. Ces actions doivent être proportionnelles au risque et rapidement communiquées à la Commission européenne et aux autres États membres. Si les mesures ne sont pas appliquées, les autorités peuvent intervenir davantage. En cas de désaccord entre États membres ou avec la Commission sur ces mesures, la Commission évalue et décide de leur justification (article 95).
Lorsqu'un état membre ou la Commission considère que le risque pour la santé et la sécurité présenté par un dispositif ne peut pas être atténué de manière satisfaisante par les mesures prises par le ou les états membres concernés, la Commission peut prendre les mesures nécessaires et dûment justifiées pour garantir la protection de la santé et de la sécurité (article 96).
Lorsque les autorités compétentes d'un état membre concluent qu'un dispositif n'est pas conforme aux exigences du Règlement mais ne présente pas un risque inacceptable pour la santé ou la sécurité, elles exigent de l'opérateur économique concerné qu'il mette un terme à la non-conformité en cause dans un délai raisonnable qu’elles définissent. Lorsque l'opérateur économique ne met pas un terme à la non-conformité dans le délai fixé, l'état membre concerné prend sans tarder toutes les mesures appropriées pour restreindre ou interdire la mise à disposition du produit sur le marché, ou pour que celui-ci soit rappelé ou retiré du marché. Il notifie sans tarder ces mesures à la Commission et aux autres états membres via le système électronique relatif à la surveillance du marché. La Commission peut définir les mesures appropriées à prendre par les autorités compétentes (article 97).
Lorsqu'un Etat membre considère que, pour protéger la santé et la sécurité, la mise à disposition sur le marché ou la mise en service d'un dispositif ou d'une catégorie ou d'un groupe spécifique de dispositifs devrait être interdite, restreinte ou assortie d'exigences particulières, ou que le dispositif ou la catégorie ou le groupe de dispositifs en question devrait être retiré du marché ou rappelé, il peut prendre toute mesure nécessaire et justifiée. L’Etat membre notifie ces mesures à la Commission et à tous les autres Etats membres via le système électronique relatif à la surveillance du marché. La Commission décide si les mesures nationales sont justifiées ou non. Lorsque l'évaluation menée par la Commission en concertation avec le GCDM et, le cas échéant, les opérateurs économiques concernés, montre que la mise à disposition sur le marché ou la mise en service d'un dispositif ou d'une catégorie ou d'un groupe spécifique de dispositifs devrait être interdite, restreinte ou assortie d'exigences particulières, ou que le dispositif ou la catégorie ou le groupe de dispositifs en question devrait être retiré du marché ou rappelé dans tous les états membres pour protéger la santé et la sécurité des patients la Commission peut adopter des actes d'exécution pour prendre les mesures nécessaires et dûment justifiées (article 98)
Les autorités compétentes des Etats membres indiquent les motifs exacts sur lesquels reposent les décisions qu’elles prennent dans le cadre de la surveillance du marché. Lorsqu'une des mesures susexposées est adressée à un opérateur économique particulier, il est informé de la décision et des voies de recours dont il dispose. L’opérateur économique doit être en mesure de présenter ses observations à l’autorité compétente dans un délai approprié. Les mesures sont immédiatement retirées ou modifiées si l’opérateur a pris des mesures correctives efficaces et que le dispositif est désormais conforme aux exigences du Règlement (article 99).
Les informations relatives à l’activité de surveillance et aux DM sont transmises via le système électronique relatif à la surveillance du marché à toutes les autorités compétentes concernées et, le cas échéant, à l'ON qui a délivré le certificat. Elles sont accessibles aux états membres et а la Commission. Les informations échangées entre états membres ne sont pas rendues publiques lorsque les activités de surveillance du marché et la coopération entre les états membres risqueraient d’en être affectées (article 100).
Le Suivi Clinique Après Commercialisation (PMCF)
(MDR 2017/745 Annex XIV Part B, article 61).
La PMCF est un suivi clinique après marquage CE consistant à recueillir des données cliniques en vie réelle, pour confirmer les revendications de performance et de sécurité d’un DM. La PMCF s’inscrit dans la PMS du fabricant. Les données de la PMCF servent donc à nourrir la PMS. Tous les dispositifs associés à un risque élevé pour le patient doivent faire l’objet d’une PMCF. La mise en place d’une PMCF est impérative lorsque :
- Les données cliniques disponibles ne couvrent pas la durée de vie du DM.
- Les données cliniques au moment du marquage CE ne sont pas extrapolables à la totalité de la population cible du fait notamment d’investigations cliniques contrôlées plus restrictives.
- Il y a des risques cliniques non maitrisés dans l’analyse des risques.
La PMCF fait partie de la documentation technique du DM. Etant un une exigence formelle de la réglementation européenne, l’absence éventuelle d’une PMCF doit être justifiée (MDR Annexe II §6.1 d).
L’Evaluation Clinique
L’article 61 §1 du MDR 271/745 indique que les fabricants « doivent planifier, réaliser et documenter une évaluation clinique » pour permettre : La confirmation de la conformité aux exigences générales pertinentes en matière de sécurité et de performances ... dans les conditions normales d'utilisation prévue du dispositif, ainsi que l'évaluation des effets secondaires indésirables et de l'acceptabilité du rapport bénéfice-risque ... doivent être fondées sur des données cliniques fournissant des preuves cliniques suffisantes
Deux documents vont être générés lors de l’évaluation clinique, le plan d’évaluation clinique (CEP) et le rapport d’évaluation clinique (CER) L’évaluation clinique doit être réalisée dans le cadre d’une demande de premier marquage CE, et est remise à jour périodiquement (tous les 1 à 5 ans en fonction de la classe de risques du dispositif et de son historique sur le marché). Cela permet ainsi de vérifier tout au long de la vie du DM sa conformité aux exigences règlementaires.
Le Plan d’Evaluation Clinique (CEP)
La planification est essentielle à un processus d’évaluation clinique efficace. Pour démontrer qu'un dispositif atteint les performances escomptées, les avantages cliniques et les spécifications de sécurité, il est nécessaire d'abord d'identifier quels sont ces paramètres. Le fabricant doit ensuite expliquer comment ces paramètres peuvent être mesurés ou démontrés (c'est-à-dire quels sont les paramètres associés) et à quoi ressemble un résultat acceptable (c'est-à-dire des références ou des mesures de performance). Pour ce faire, il est nécessaire de commencer par une description claire et suffisamment détaillée de l’usage prévu du dispositif, ainsi que par une spécification des normes de soins pour cet usage prévu (autrement appelé « état de l’art »).
Le Rapport d’évaluation Clinique (CER)
Un rapport d'évaluation clinique (CER) documente les conclusions d'une évaluation clinique et les preuves cliniques d'un dispositif médical. Un CER peut contenir des données cliniques provenant de :
- Investigation clinique du dispositif en cours d'évaluation.
- Investigation clinique ou autres études rapportées dans la littérature scientifique pour un dispositif équivalent.
- Littérature scientifique évaluée par des pairs faisant état d'autres expériences cliniques du dispositif évalué ou d'un dispositif équivalent.
- Données de surveillance post-commercialisation (PMS) cliniquement pertinentes avec un accent particulier sur le suivi clinique post-commercialisation (PMCF).
Le CER vise à démontrer qu'il existe suffisamment de preuves cliniques pour confirmer la sécurité et les performances, y compris les avantages cliniques, du dispositif évalué lorsqu'il est utilisé comme prévu. Les CER sont requis pour tous les dispositifs médicaux dans l'UE et font partie de la documentation technique du règlement sur les dispositifs médicaux MDR 2017/745.
Synthèse
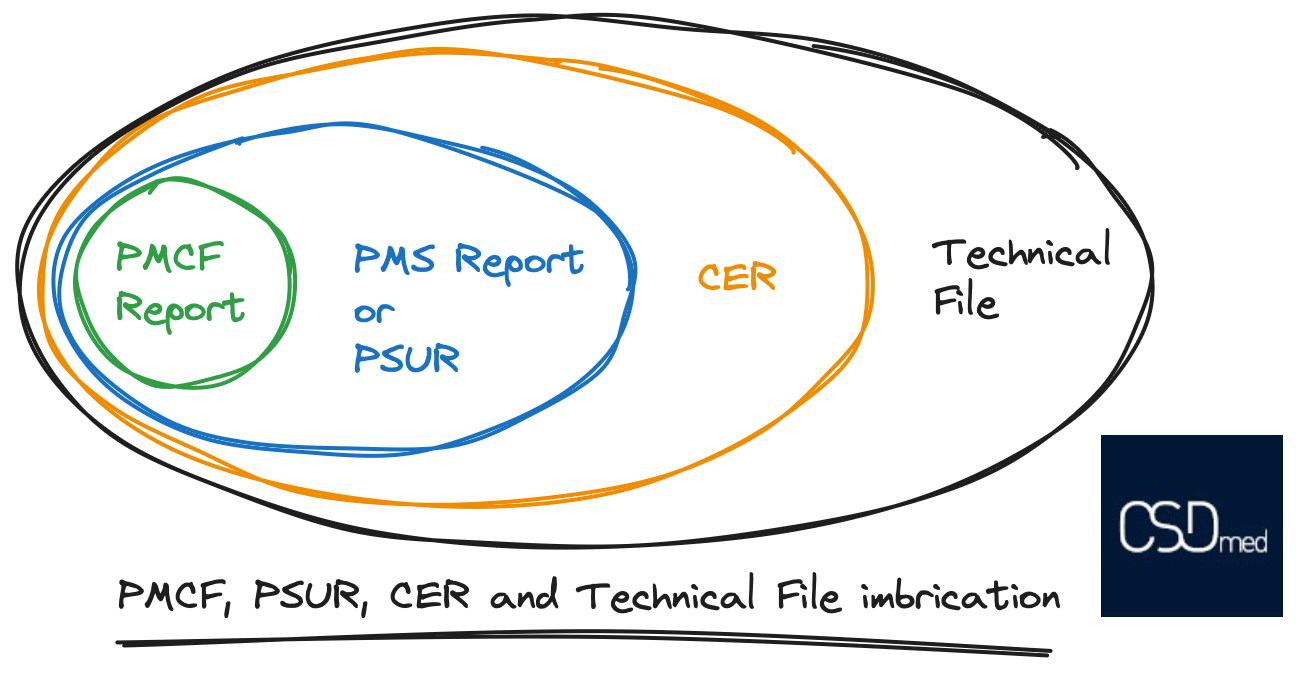
Une image vaut mieux que mille mots, voici donc une illustration expliquant les imbrications entre les différents éléments, du Rapport de Suivi Clinique Après Commercialisation (PMCF), au Rapport de Surveillance Après Commercialisation (PMSR) ou au Rapport Périodique Actualisé de Sécurité (PSUR), au rapport d’Evaluation Clinique (CER), pour finir au Dossier Technique (TF). Les éléments s’imbriquent comme dans une poupée russe.
Vous pouvez également consulter notre article sur le contenu du dossier technique de marquage CE.
Nous sommes à votre service
Le Règlement Européen MDR 2017/745 a ajouté de nouvelles exigences et celles-ci font partie de la documentation à présenter à l’organisme notifié (par exemple revue de l’évaluation clinique poussée, procédure de PMS, résultats de CQ issus de la validation des produits, preuves de compétences du personnel, etc.).
CSDmed apporte son expertise et une approche méthodique à ses clients, start-ups, fabricants, importateurs et distributeurs de dispositifs médicaux, grâce à une équipe d’experts et de consultants spécialisés, qui pourront traiter la transition MDR dans son entièreté.
🔗 Contactez-nous et découvrez comment nous pouvons vous aider.